generalitate
Sindromul Noonan este o boală genetică rară, uneori ereditară, care modifică dezvoltarea normală a diferitelor părți anatomice ale corpului. Caracteristicile neobișnuite ale feței, statura scurtă și unele defecte cardiace congenitale reprezintă principalele semne patologice și diagnostice.

Consecințele bolii nu sunt întotdeauna dramatice: în unele cazuri, de fapt, sindromul Noonan poate provoca o simptomatologie nuanțată și poate permite o viață normală.
Scurtă referire la genetică: ADN și cromozomi
Înainte de a descrie sindromul Noonan, este bine să faceți o scurtă referire la genetică.
CHROMOSOME ȘI ADN
Fiecare celulă a unei ființe umane sănătoase are 23 de perechi de cromozomi omologi : 23 sunt materni, adică moșteniți de la mamă, iar 23 sunt paterni sau moșteniți de la tată. Câteva dintre aceste cromozomi sunt sexuale, adică determină sexul individului; restul de 22 de perechi, pe de altă parte, sunt compuse din cromozomi autozomali . În totalitate, cele 46 de cromozomi umane conțin întregul material genetic, mai bine cunoscut sub numele de ADN . În ADN-ul unui individ sunt scrise trăsăturile sale somatice, predispozițiile sale, darurile sale fizice și așa mai departe.
GENE ȘI MUTAȚII ADN
ADN-ul este organizat în mai multe secvențe, mai mult sau mai puțin lungi, numite gene . Fiecare genă ocupă un cromozom specific și omologul său, așa cum este prezent în două exemplare numite alele . O alelă provine de la mamă și se află în cromozomul matern; cealaltă alelă vine de la tată și este adăpostită în cromozomul paternal.

Figura: organizarea unei gene în interiorul unei perechi de cromozomi omologi. O pereche de cromozomi omologi conține gene specifice, toate având două variante, alele, ocupând aceeași poziție cromozomală și realizând aceleași funcții (cu excepția mutațiilor). Perechea de cromozomi din stânga are două alele egale (ambele albastru deschis); pe de altă parte, perechea dreaptă are două alele diferite (una este roșie, cealaltă este albastră).
Din gene generează proteine prezente în corpul nostru. Atunci când apare o mutație ADN, o genă (de obicei, o alelă) a unui cromozom dat poate fi defectă și poate produce, prin urmare, o proteină defectă.
Ce este sindromul Noonan?
Sindromul Noonan este o boală genetică, uneori ereditară, care determină diferite anomalii anatomice (și nu numai) în multe părți ale corpului. Principalele modificări apar la nivelul feței, inimii și scheletului, dar nu este exclus ca sistemul reproductiv, sistemul limfatic, pielea, sistemul nervos, ochii, urechile, sângele și rinichii.
Anomaliile care caracterizează sindromul Noonan nu sunt întotdeauna aceleași la toți pacienții: la cineva ei sunt necunoscuți, astfel încât să meargă aproape neobservată și să permită o viață aproape normală; la altcineva, pe de altă parte, ele sunt atât de accentuate încât pun purtătorul în pericol grav de viață.
Epidemiologie
Sindromul lui Noonan este mai puțin frecvente: afectează, de fapt, un copil pentru fiecare 2500 de nou-născuți. Atât bărbații, cât și femeile sunt afectați în mod egal și nu există nici un grup etnic mai predispus decât alții.
cauze
Sindromul Noonan este o boală genetică, deci este cauzată de o schimbare a ADN-ului . Cercetătorii sunt destul de siguri că genele implicate sunt cel puțin opt, dar încă nu pot explica în mod clar cum provoacă anomalii anatomice.
Ce fac aceste opt gene?
ROLUL
Genele implicate în sindromul Noonan au un rol fiziologic deosebit: ele produc proteine care permit creșterea și dezvoltarea celulelor noastre. Cu alte cuvinte, aceste proteine sunt scopuri de reglementare, care îngrijesc în detaliu viața și soarta fiecărei celule.
Când genele care creează aceste proteine se modifică, mecanismele de reglare a celulelor descrise mai sus sunt, de asemenea, modificate, cu efecte grave asupra organismului.
GENELE IMPLICITE
Dintre cele opt gene implicate în sindromul Noonan, doar patru au fost identificate pe deplin, deoarece mută la o frecvență statistic semnificativă.
Acestea sunt:
- PTPN11 : mutația sa reprezintă aproximativ 50% din cazuri. Se află în cromozomul 12, iar descoperirea implicării sale în sindromul Noonan datează din 2001. Proteina pe care o produce are un rol fundamental în dezvoltarea embrionară: guvernează, de fapt, creșterea, diferențierea și divizarea celulelor, acelea a inimii în particular. Stim cu siguranta ca o alela mutanta este suficienta pentru ca boala sa se manifeste (dominanta).
- SOS1 : sa schimbat în 10-15% din cazuri. Sa înțeles că el ar putea fi implicat în boală în 2006.
- RAF1 : sa modificat în 5-10% din cazuri. Am aflat doar despre rolul său în 2007.
- KRAS : mutația sa reprezintă aproximativ 2% din cazuri. Descoperirea implicațiilor sale este recentă: 2006.
După cum vedeți, cercetarea referitoare la aceste gene este destul de recentă și acest lucru explică de ce mai există puncte în așteptare.
HEREDITARITATEA: DA? SAU NU?
Mutațiile genetice, în 50% din cazuri, sunt sporadice (adică datorită hazardului), în timp ce în restul de 50% acestea sunt transmise de unul dintre părinți .
De obicei, acei părinți care transmit boala copiilor lor nu știu că suferă de sindromul lui Noonan, pentru că, probabil, aceasta nu este o formă gravă și evidentă.
simptomele
Sindromul lui Noonan se manifestă cu câteva semne caracteristice în mai multe părți ale corpului.
Aceste semne constau în anomalii anatomice mai mult sau mai puțin marcate și în condiții patologice mai mult sau mai puțin grave. De fapt, unii pacienți manifestă boala în mod clar, din toate punctele de vedere; altele, pe de altă parte, se prezintă cu o simptomatologie mai nuanțată sau se limitează la anumite locații.
CARACTERISTICILE PATOLOGICE PRINCIPALE ȘI SECUNDARE
Trăsături neobișnuite ale feței, statură scurtă și o serie de defecte cardiace congenitale sunt anomaliile găsite la toți cei care suferă de sindromul Noonan; din acest motiv, sunt luate în considerare principalele caracteristici ale bolii (NB: termenul congenital înseamnă prezența de la naștere ).
Pe de altă parte, anomaliile secundare sunt considerate caracteristici secundare, mai puțin comune decât cele precedente:
- Afișarea ușoară a hematoamelor și a hemoragiilor
- Dificultăți de învățare și anomalii comportamentale
- Deficiențe vizuale de diferite tipuri
- limfedemul
- Hipotonia musculară
- Pierderea auzului
- Infertilitatea și aparatul genital anormal
- Alimentație dificilă (în copilărie)
- Anomalii scheletice
FACE DE FACE
Caracteristicile anormale ale feței pot fi observate deja atunci când pacientul este foarte mic; apoi, cu timpul, se observă variația lor, devenind permanentă la vârsta adultă.
- Copilăria foarte fragedă (mai puțin de un an): ochii sunt îndepărtați unul de celălalt și tind spre partea de jos. Urechile sunt mici și orientate spre partea din spate a capului. Brazda, prezentă deasupra buzei superioare, este adâncă; gâtul este scurt și linia de păr din spatele capului este scăzută.
- Copilăria : ochii încep să devină proeminenți, în timp ce pleoapele sunt coborâte ( ptoză ) și devin mai îngroșate. Nasul este zdrobit la rădăcină, dar are o bază lată și un vârf bulbos.
- Copilăria : la caracteristicile anterioare, o inexpresivitate a feței, buzele mărită și o lungime mai mare a feței sunt adăugate.
- Adolescenta : fruntea devine largă și bărbia devine ascuțită, făcând astfel chipul să pară un triunghi. Unele caracteristici faciale sunt accentuate, ca liniile care încep de la nas și ajung la colțurile gurii, în timp ce ochii devin mai puțin proeminenți. Gâtul scurt este îmbogățit cu alte detalii: apar primele pliuri ( pterigium colli ) și mușchii trapezius sunt mai voluminoase.
- Vârsta adulților : brazdele care curg de la nas până la laturile gurii sunt profunde și evidente. Gura devine și mai proeminentă și pielea devine șifonată și clară. Ochii sunt întotdeauna la distanță și cu pleoapele îngroșate și afectate de ptoză.
STAREA LUMINĂ
Greutatea și lungimea la naștere sunt normale. Cu toate acestea, în primele 18-24 luni, se observă că creșterea este lentă și întârziată, în comparație cu copiii de aceeași vârstă.

Figura: fața unui adult cu sindromul Noonan. De pe site: login.aafp.org
Acest lucru se datorează unei probleme hormonale ( hormon de creștere, GH ), dar și anumitor dificultăți care îi prezintă copilului în a mânca.
Întârzierea creșterii se observă și în timpul copilăriei și, mai ales, în timpul pubertății, când băieții au tendința de a se dezvolta brusc. Diferențele față de colegi, în acest moment al vieții, sunt evidente.
În absența tratamentului, înălțimea la bărbați atinge o medie de 162 cm, în timp ce la femele atinge 153 cm. Cu ajutorul tratamentelor terapeutice corecte, înălțimea poate fi inclusă și în mediul normal.
DEFECTELE CARDIACULUI CONGENITAL
80% dintre pacienții cu sindrom Noonan se nasc cu defecte cardiace mai mult sau mai puțin severe. Aceste anomalii pot fi:
- Stenoza valvei pulmonare : aceasta este o îngustare (stenoză) a supapei inimii (care permite sângelui să curgă de la ventriculul drept al inimii la artera pulmonară). Artera pulmonară este responsabilă pentru transportul sângelui în plămâni, pentru oxigenarea sa.
- Stenoza arterelor pulmonare : este o îngustare a arterei pulmonare; acest defect, ca cel precedent, limitează cantitatea de sânge care ajunge la plămâni pentru oxigenare.
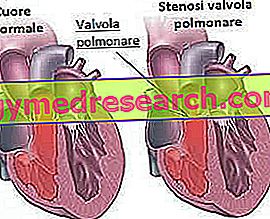
- Defectul septal inter-ventricular (între ventricule) sau septul inter-atrial (între atriu): constă în formarea unei găuri între peretele de țesut care separă ventriculii (sau cel care separă atria). Acest lucru face ca sangele sa curga intre cele doua compartimente, provocand probleme circulatorii, uneori chiar grave.

ALTE ANOMALII
Deoarece acestea sunt numeroase manifestări, vom încerca să le descriem în principalele lor trăsături.
- Dificultăți de învățare și anomalii comportamentale . Inteligența poate fi sub media. Cu toate acestea, există mulți indivizi care, în ciuda faptului că sunt purtători ai bolii, au un IQ normal.
Același lucru este valabil și pentru comportament: în unele cazuri, pacientul este iritabil, repetă sunetele și cuvintele altor persoane și are nevoi nutriționale ciudate.
- Afișarea ușoară a hematoamelor și a hemoragiilor . La 50% dintre pacienți, capacitățile de coagulare sunt reduse. Prin urmare, pierderile de sânge, chiar și după traumatismele minore, sunt vizibile. Este o caracteristică de a nu fi neglijat când se iau anumite medicamente (aspirină) sau se efectuează intervenții chirurgicale (toate, de la cele mai invazive la extragerea simplă a unui dinte).
- Deficiențe vizuale de diferite tipuri . Aproximativ jumătate dintre pacienți prezintă probleme de vedere de diferite tipuri. Strabismul, astigmatismul, miopia, ochiul leneș ( ambliopia ), hipermetropia și nistagmusul sunt posibilele defecte vizuale care pot apărea la pacienții cu sindrom Noonan.
- Lymphedema . Este un defect la nivelul sistemului limfatic . Fluidul limfatic, sau limfa, se acumulează în unele părți ale corpului, cum ar fi mâinile și picioarele. Apare în special în timpul copilăriei.
- Hipotonia musculară . Este o reducere a tonusului muscular. Este tipic pentru copilărie și copilărie.
- Pierderea auzului . Este un episod temporar, în vârstă fragedă. Aceasta se datorează infecțiilor frecvente ale urechii medii.
- Infertilitatea și aparatul genital anormal . Tulburările sistemului genital sunt observate la 60% dintre pacienți. Adesea, subiecții sunt bărbați care suferă de criptorhidism (adică lipsa coborârii unuia sau a ambilor testiculi în scrot). Pentru a rezolva această problemă, trebuie utilizată o intervenție chirurgicală. În caz contrar, fără intervenție, individul are un număr redus de spermatozoizi, deci este mai puțin fertil.
Femeile cu sindromul Noonan nu au de obicei aceste probleme.
- Hrănire dificilă . Este o problemă tipică a copilăriei și contribuie la întârzierea creșterii. Copilul are probleme cu alăptarea laptelui matern și tinde să vomite după fiecare masă.
- Anomalii scheletice . În special la tinerii adolescenți, pot fi observate hipermobilități articulare (adică articulații cu o gamă largă de mișcări), scolioză, excavatum toracic (sau pâlnie), piept și șuvițe carinați distanțate unul de celălalt într-un mod neobișnuit.
Când și ce ar trebui să vă dați dovadă unui doctor?
Dacă observați, la copil, caracteristici faciale, cum ar fi cele descrise anterior, se recomandă supunerea copilului la un examen pediatric. Medicul va efectua evaluările corespunzătoare și, dacă suspectează sindromul Noonan, va efectua testele de diagnostic necesare.
În unele cazuri, anomaliile anatomice sunt neclare, astfel încât cei care poartă boala nu știu că sunt afectați. Cu toate acestea, chiar și în aceste situații, diagnosticul este important, din cauza problemelor cardiace care pot apărea.
complicaţiile
Complicațiile legate de sindromul Noonan depind de gravitatea bolii. Prin urmare, din acest punct de vedere, fiecare pacient reprezintă un caz în sine.
Printre complicațiile principale, merită un citat:
- Sarcina intelectuală serioasă
- Extrem de compromis capacitățile de coagulare a sângelui
- Formarea de limfedem în jurul organelor vitale, cum ar fi inima și plămânii
- Anomaliile anatomice severe ale sistemului genital, cu repercusiuni asupra celei urinare (în particular rinichii)
diagnostic
O patologie cum ar fi sindromul Noonan, caracterizată printr-un număr enorm de semne speciale, poate fi diagnosticată chiar și după o examinare obiectivă . Cu toate acestea, în unele cazuri, boala nu este foarte evidentă, atât de mult încât, așa cum sa spus deja în mai multe rânduri, poate trece neobservată până la pubertate sau la vârsta adultă (când apar nevoile sexuale ale pacientului).
În orice caz, pentru a clarifica orice îndoială, există teste genetice foarte fiabile, cum ar fi așa-numitul cariotip .
Odată ce boala a fost diagnosticată, este important să se monitorizeze, prin intermediul unor teste specifice, condițiile patologice potențial periculoase pentru pacient, cum ar fi defectele cardiace sau capacitățile reduse de coagulare. Pe baza rezultatelor acestor teste, gravitatea fiecărui caz considerat poate fi stabilită cu exactitate.
Semnele cele mai importante pentru examenul obiectiv:
| Monitorizarea este efectuată de:
|
tratament
Fiind o boală genetică, sindromul lui Noonan nu este vindecător.
Cu toate acestea, simptomatologia pe care o produce poate fi limitată și atenuată de terapii destul de eficiente.
ÎNGRIJIREA DE PROBLEME CARDIAC
Știind care sunt tulburările care afectează inima este esențială pentru a putea stabili cea mai potrivită terapie.
Anumite anomalii cardiace pot fi controlate printr-un tratament farmacologic simplu (diuretice, antiaritmice și beta-blocante); altele mai severe (de exemplu, stenoza valvei pulmonare sau defectele septului) pot necesita intervenții chirurgicale.
În orice caz, monitorizarea periodică este esențială, deoarece o situație aparent inofensivă se poate transforma într-o evoluție foarte gravă și dramatică.
CURSURI HORMONALE PENTRU CREȘTERE
Adesea, dezvoltarea redusă a stării se datorează producției rare de hormon de creștere sau GH . Prin urmare, administrarea exogenă a unuia dintre analogii săi ( somatropină ), creată în laborator, are efecte excelente, cu condiția ca programul terapeutic să fie respectat cu scrupulozitate. Acesta din urmă este foarte simplu: injecții zilnice, începând cu 3-5 ani de viață.
Efectele secundare sunt rare și constau în majoritatea de mâncărime și roșeață în zona de injectare.
Sunt recomandate verificări periodice ale nivelelor hormonale.
ÎNGRIJIREA DEFICITULUI DE ÎNVĂȚĂMÂNT
În unele cazuri, sindromul lui Noonan poate afecta considerabil abilitățile intelectuale. În aceste situații, pacientul are nevoie de un sprijin valabil, în special la școală.
ALTE TRATAMENTE
Pentru defectele vizuale pot fi suficiente ochelari adecvați pentru patologia diagnosticată; este rar să recurgă la intervenții chirurgicale.
În ceea ce privește problemele de coagulare, există medicamente care promovează coagularea, care trebuie administrate atunci când este necesar. Mai mult, o recomandare de reținut nu este să luați medicamente anticoagulante, cum ar fi aspirina și derivații săi.
Dacă limfedemul se formează în jurul unor organe critice, cum ar fi inima sau plămânii, este posibilă scurgerea fluidului limfatic prin introducerea unui tub special. Chirurgia, în aceste cazuri, este o eventualitate rară.
În cele din urmă, în ceea ce privește infertilitatea și defectele sistemului genital, criptorchidismul poate fi rezolvat printr-o intervenție specifică (NB: cititorul reaminteste că infertilitatea, în sindromul Noonan, este o problemă tipic masculină).
prognoză
Prognoza variază de la pacient la pacient. La unele persoane, de fapt, sindromul Noonan provoacă o simptomatologie nuanțată și permite o viață aproape normală (cu condiția să se aplice o îngrijire adecvată); în altele, cu toate acestea, compromite serios abilitățile intelectuale și starea generală a sănătății.
Prin urmare, prognosticul poate fi pozitiv (primul caz), dar și negativ (al doilea caz).
Pe lângă aceste două situații extreme, se introduc cazuri moderate, adică căile de mijloc între formele grave și cele grave. Confruntată cu aceste circumstanțe, pentru ca prognosticul să fie pozitiv, este esențial să se stabilească un diagnostic precoce urmat de un tratament adecvat și la fel de precoce. Doar în acest fel prognosticul poate avea un impact pozitiv.
PREVENIREA
Prevenirea mutațiilor sporadice nu este, din păcate, posibilă. Formele ereditare, pe de altă parte, pot fi prevenite prin informarea persoanelor fertile care suferă de sindromul Noonan de posibilitatea transmiterii bolii la copiii lor.