generalitate
Craniosinostoza este termenul prin care medicii indică o anomalie a craniului datorată fuziunii prematură a unuia sau mai multor suturi craniene.
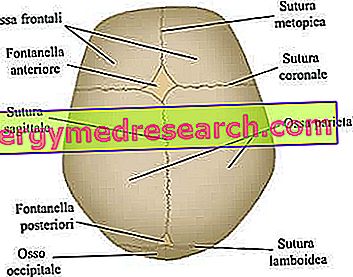
Suturile craniene sunt articulațiile fibroase care unesc oasele bolii craniene (oasele frontale, temporale, parietale și occipitate).
Craniosinostoza poate fi un fenomen izolat (craniosinostoza non-sindromică) sau rezultatul unor afecțiuni morbide deosebite (craniosinostoza sindromică). Printre condițiile morbide care determină fuziunea prematură a suturilor craniene, cele mai cunoscute sunt: sindromul Crouzon și sindromul Apert.
Cu fuziunea prematură a suturilor craniene, structurile encefalice nu dispun de spațiul adecvat pentru creștere. Acest lucru are consecințe diferite, incluzând în principal creșterea presiunii intracraniene (hipertensiunea intracraniană).
Diagnosticarea în timp util și precis permite planificarea unui tratament ad-hoc . Acesta din urmă are un tip chirurgical și are ca obiectiv final separarea timpurie a suturilor fusely.
Reamintește despre anatomia craniului uman
Furnizat cu oase și cartilaje, craniul este structura scheletică a capului care constituie fața și protejează creierul, cerebelul, trunchiul cerebral și organele senzoriale.

Pentru a simplifica studiul si intelegerea craniului, anatomii s-au gandit sa-l imparta in doua compartimente, numite neurocranium si splancnocranium .
neurocraniu
Neurocraniul este regiunea craniană superioară care conține creierul și câteva organe senzoriale principale. Oasele sale cele mai importante - strict plate - sunt oasele frontale, temporale, parietale și occipitate; acestea, împreună, formează așa-numita boltă craniană .
splanchnocranium
Splanchnocraniul, sau masivul facial, este regiunea antero-inferioară a craniului, compusă din oase egale și inegale. Ea reprezintă structura scheletului feței, prin urmare ea conține elemente osoase cum ar fi maxilarul, maxilarul superior, pomeții, osul nazal etc.
Ce este craniosinostoza?
Craniosynostoza este o anomalie rară a craniului, caracterizată printr-o formă a capului nefiresc din cauza fuziunii prematură a unuia sau a mai multor suturi craniene . Suturile craniene sunt articulațiile fibroase care unesc oasele bolii craniene (oasele frontale, temporale, parietale și occipitate).

De pe site: //www.wkomsi.com/
CÂND trebuie să aibă loc închiderea suturilor craniene?
În condiții normale, fuziunea suturilor craniene are loc în perioada postnatală (NB: unele procese se termină chiar la vârsta de 20 de ani). Acest lung proces de fuziune permite creierului să crească și să se dezvolte în mod corespunzător.
Dacă, cum se întâmplă în cazul craniosinostozei, fuziunea are loc prea devreme - prin urmare, în timpul vieții prenatale, perinatale sau timpurii din copilărie - elementele encefalice (creierul, cerebelul și trunchiul cerebral) și unele organe de simț (ochii în particular) o modificare a formei și a creșterii.
cauze
Procesul fiziopatologic care determină craniosinostoza este fuziunea prematură a suturilor craniene .
Acest proces poate reprezenta un fenomen izolat - în cazul în care "izolat" înseamnă că nu este asociat cu nici o stare morbidă particulară - sau poate fi consecința unor anumite sindroame, aproape întotdeauna de natură genetică.
În lumina acestui fapt, medicii au considerat că clasifică craniosinostoza în două categorii:
- Craniosinostoza non-sindromică . Termenul non-sindrom indică faptul că anomalia craniană nu este asociată cu nici o patologie sau alt defect fizic.
- Craniosinostoza sindromică . Termenul de sindrom înseamnă că malformația craniană este rezultatul unui anumit sindrom, în majoritatea cazurilor de tip genetic.
CRANIOSINOSTAZA NON-SYNDROMICĂ
Doctorii și cercetătorii nu au stabilit încă cauzele craniosinostozei non-sindromice.
Ei au propus diverse ipoteze - inclusiv influența factorilor de mediu sau a problemelor asemănătoare hormonilor - dar niciuna dintre aceste teorii nu a fost confirmată prin rezultate experimentale.
Prin urmare, pentru a înțelege originea exactă a anomaliei, sunt necesare studii suplimentare.
CRANIOSINOSTOZA SINDROMICA
Conform ultimelor cercetări medicale, există mai mult de 150 de sindroame diferite, toate foarte rare, capabile să provoace craniosinostoză.
Printre aceste sindroame, cele mai cunoscute și comune sunt:
- Sindromul Crouzon . Rezultatul mutațiilor specifice în genele FGFR2 (cromozomul 10) și FGFR3 (cromozomul 4), această afecțiune morbidă particulară afectează un nou-născut la fiecare 60 000 și implică prezența anomaliilor exclusiv la nivelul capului și al feței.
- Sindromul Apert . Apare în principal din cauza mutațiilor genei FGFR2 (la fel ca sindromul Crouzon) și afectează un nou-născut la fiecare 100 000 sau cam asa ceva.
Spre deosebire de sindromul Crouzon, modificările genetice ale FGFR2 sunt de așa natură încât malformațiile implică nu numai craniul și fața, ci și mâinile și picioarele.
- Sindromul Pfeiffer . Aceasta apare din cauza mutațiilor în gena "obișnuită" a FGFR2 și o genă cu funcții similare, numită FGFR1 (cromozomul 8). Particularitatea acestor mutații este că - în afară de deformările craniului și feței - ele determină de asemenea: sindactilă, brachydactyly și degetele mari și degetele de la picioare (disproporționate față de celelalte degete).
Sindromul Pfeiffer are o incidență de 1 din 10000 de nou-născuți.
- Sindromul Saethre-Chotzen . Este o afecțiune genetică care afectează un nou-născut la fiecare 50 000 sau cam asa ceva. Cauzează diverse malformații la nivelul craniului, feței, mâinilor și picioarelor. Unele mutații specifice ale genei TWIST1, localizate pe cromozomul 7, sunt responsabile pentru apariția sindromului Saethre-Chotzen.
EPIDEMIOLOGIA CRANIOSINOSTEZEI
Conform celor mai recente statistici, se pare că un copil de la aproximativ 1800-3000 suferă de craniosinostoză.
În ceea ce privește sexul cel mai afectat, mai multe studii clinice au arătat că 3 din 4 pacienți sunt bărbați. Motivul pentru care craniosinostoza este mai răspândită la populația masculină este complet necunoscută.
Factori de risc de craniosinostoză.
- Greutate mică la naștere
- Nașterea prematură
- Varsta paterna avansata
- Fumatul matern în timpul sarcinii
Simptome și complicații
Majoritatea simptomelor care pot fi observate în prezența craniosinostozei se datorează creșterii presiunii în craniu . În medicină, creșterea presiunii în craniu se numește hipertensiune intracraniană sau hipertensiune intracraniană .
În prezența craniosinostozei, hipertensiunea intracraniană este o consecință a faptului că creierul și alte structuri din interiorul craniului nu dispun de spațiul potrivit pentru a crește, astfel că ele se duc la împingerea structurilor osoase ale capului.
Acestea fiind spuse, este important să rețineți că, dacă suturile craniene implicate sunt multe sau dacă condiția nu este tratată în timp, craniosinostoza poate duce la o dezvoltare redusă a abilităților cognitive și la un IQ scăzut.
SIMPTOME A HIPERTENSIUNII ENDOCRANICE
Simptomele posibile ale hipertensiunii intracraniene sunt:
- Durere de cap persistentă. De obicei se agravează dimineața și noaptea.
- Probleme de vedere. Acestea constau în viziune dublă, vedere încețoșată și vedere încețoșată.
- vărsături
- iritabilitate
- Ochii umflați sau proeminenți
- Dificultate în urmărirea mișcării obiectelor
- Probleme de auz
- Probleme respiratorii
- Modificări ale stării mentale
- papilledema
Numărul de suturi craniene implicate în dezvoltarea craniosinostozei are o influență semnificativă asupra prezenței hipertensiunii intracraniene.
De exemplu, medicii au observat că implicarea unei singure suturi craniene induce hipertensiune intracraniană la un procent de 15% dintre pacienți; în timp ce implicarea a cel puțin două suturi duce la o creștere a presiunii în craniu la cel puțin 60% dintre pacienți.
În prezența unei forme ușoare de craniosinostoză, hipertensiunea endocraniană începe să fie problematică, provocând simptomatologia menționată anterior, în jur de 4-8 ani de viață.
SEMNELE CRANIOSINOSTEZEI
Printre semnele de craniosinostoză, cele mai frecvente sunt:- Formări de crestături rigide de-a lungul suturilor craniene
- Anormalități la nivelul fontanelor craniene
- Cap cu dimensiuni care nu sunt proporționale cu restul corpului
TIPURI DE CRANIOSINOSTOZĂ
Forma capului pacienților cu craniosinostoză depinde de ce suturi craniene s-au închis prematur.
După ce au observat acest lucru, medicii au considerat oportun să distingă craniosinostoza în diferite tipuri, în funcție de suturile craniene implicate.
Tipurile de craniosinostoză sunt:
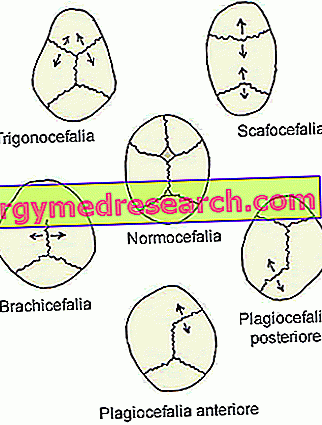
- Sajoză sagitală ( dolichocephaly sau scafocephaly ). Este cel mai frecvent tip de craniosinostoză; în fapt, ea caracterizează aproximativ jumătate din cazurile clinice.
Prezența sa coincide cu închiderea prematură a suturilor craniene sagitale, situate în partea superioară a craniului, între oasele parietale.
De la //en.wikipedia.org/wiki/Plagiocephaly
- Craniosinostoza coronală ( brachycephaly ) Este al doilea tip de craniosinostoză cel mai frecvent; prezintă câte un caz clinic la fiecare patru.
Debutul său implică fuziunea prematură a suturilor coronale, care se desfășoară între osul frontal și oasele parietale.
- Metastazare ( trigonocefalie ). Este un tip destul de rar de craniosinostoză, care distinge doar 4-10% din cazuri.
Aspectul său coincide cu fuziunea prematură a suturii metopice (sau frontale), care curge de la nas până la vârful capului, separând osul frontal în două. În mod obișnuit, această sutură se consolă în mod natural în cel de-al șaselea an de viață.
- Lăcusta sinoboidală ( plagiocefalie ). Este cel mai rar tip de craniosinostoză. De fapt, aceasta distinge doar 2-4% din cazurile clinice.
Prezența sa implică fuziunea timpurie a suturii lambdoid, situată între oasele parietale și osul occipital, la partea din spate a capului.

COMPLICAȚII
Pe lângă faptul că afectează dezvoltarea intelectuală, craniosinostoza netratată poate determina:
- Așa-numitul sindrom de apnee în somn obstructiv .
- Modificări permanente ale feței la nivelul ochiului și urechii în special.
- Deformările permanente la baza craniului (de exemplu, malformația sau sindromul lui Arnold-Chiari).
Principalele suturi craniene implicate în procesul craniosinostozei. De pe site-ul: www.sciencebasedmedicine.org
- Hidrocefalie .

diagnostic
Pentru a diagnostica craniosinostoza, examinarea obiectivă, evaluarea antecedentelor clinice și a imaginilor radiologice furnizate prin raze X sau scanarea CT la nivelul capului sunt esențiale.
Dacă craniosinostoza a fost de tip sindromic, este de asemenea important să se stabilească condiția morbidă care a determinat debutul ei. Prin urmare, medicii ar putea recurge la analize de sânge și, mai presus de toate, la consiliere genetică .
OBIECTIV EXAM
Examinarea obiectivă constă în analizarea atentă de către medic a semnelor clinice prezente pe capul subiectului suspectat de a suferi de craniosinostoză.
În general, medicul pediatru este implicat în acest diagnostic important de diagnosticare.
ISTORIA CLINICĂ
Evaluarea istoricului clinic este importantă pentru diagnosticare, deoarece include întrebări legate de factorii de risc ai craniosinostozei.
Deci, medicul (de obicei, întotdeauna un pediatru) va investiga dacă:
- Copilul sa născut prematur sau cu greutate mică.
- Care era vârsta tatălui în momentul concepției?
- Dacă mama a fumat în timpul sarcinii.
EXAMINAREA RADIOLOGICĂ
X-ray-ul și scanarea CT la cap servesc mai mult decât orice altceva pentru a confirma diagnosticul și pentru a arăta medicului care suturi craniene s-au topit prematur.
Cunoașterea suturilor craniene implicate permite planificarea celui mai potrivit tratament chirurgical.
tratament
Craniosinostoza este vindecabilă numai prin intervenție chirurgicală .
Aceasta din urmă constă într-o operațiune de separare a suturilor fusel craniene între ele în mod precoce.
Scopul terapeutic final al intervenției chirurgicale este acela de a oferi structurilor creierului și unor organe de simț, cum ar fi ochii, spațiul necesar pentru a se dezvolta și a funcționa la maximum.
Cel mai bun moment pentru a interveni
Nu există nici un acord total din partea medicilor cu privire la cel mai bun moment pentru a efectua operația de craniosinostoză.
Potrivit unor experți, perioada ideală pentru operație ar fi în copilărie târziu, când riscul de recidivă (adică o a doua fuziune prematură a suturilor craniene) este mai mic. În cazul reapariției, intervenția trebuie repetată și acest lucru nu este recomandat, având în vedere delicatețea procedurii.
Potrivit altor experți, timpul cel mai potrivit ar fi fost în copilăria timpurie (între 6 și 12 luni de viață), când craniul nu este încă complet osificat și oasele sunt încă modelate. Posibilitatea de modelare a oaselor (maleabilitate) permite rezolvarea oricăror anomalii morfologice ale oaselor, care ar putea provoca defecte estetice grave și probleme funcționale (la maxilar sau la ochi) la o vârstă mai matură.
POSIBILE DE ABORDARE SURGICALĂ
Există două abordări chirurgicale diferite: chirurgia tradițională, denumită și "aer liber", și chirurgie endoscopică.
- Chirurgie tradițională (sau "aer liber") .
Aceasta implică anestezie generală (astfel încât pacientul este inconștient în timpul întregii proceduri) și practicarea unei incizii chirurgicale pe cap, exact unde imaginile radiologice au arătat anomalia craniană.
Prin incizia pe cap, chirurgul chirurgical (neurochirurg) îndepărtează osul anormal și îl încredințează unui specialist în chirurgia craniofaccială, care îl modifică și îi conferă o formă care permite dezvoltarea normală a structurilor encefalice.
După modificare, neurochirurgul înlocuiește osul în poziția inițială și închide incizia cu suturi.
La fel ca multe intervenții chirurgicale tradiționale, operația "cerului deschis" este mai degrabă invazivă; cu toate acestea, este avantajos să puteți modifica structura osoasă într-un mod precis și cu rezultate bune.
- Chirurgie endoscopică .
Aceasta implică utilizarea unui endoscop, a unui instrument similar unui tub flexibil, echipat cu o cameră cu fibră optică la un capăt și conectat la un monitor.
Din punct de vedere operativ, constă în introducerea endoscopului într-o deschidere făcută pe craniu și în separarea, prin intermediul endoscopului în sine, a suturii topite (sutură).
Neurochirurgul reușește să se orienteze în interiorul capului, datorită imaginilor pe care camera le proiectează pe monitorul conectat extern.
Chirurgia endoscopică este considerabil mai puțin invazivă decât operația "open field" (chiar și cea mai scurtă perioadă de spitalizare), însă are două dezavantaje: este indicată doar pentru pacienții de câteva luni (6 în general) care posedă oase care încă pot fi modelate; este la risc mai mare de recidivă.
Faza POST-OPERATIVĂ
De obicei, un pacient cu craniosinostoză, care a suferit o intervenție chirurgicală, trebuie să rămână în spital timp de aproximativ 4-5 zile după operație. În acest timp, neurochirurgul și membrii personalului său monitorizează periodic parametrii vitali și verifică dacă totul se desfășoară fără probleme.
După demisie, se prevede o serie de controale periodice, care sunt, la început, semestriale și apoi, odată cu creșterea pacientului, anual.
prognoză
Prognoza depinde de diferiți factori, printre care:
- Cauze care au provocat craniosinostoza. Unele boli genetice responsabile pentru această anomalie sunt foarte grave și din cauza prognozei sărace.
- Poziția fuselului se sutură prematur. Dacă suturile se află în poziții care, pentru neurochirurg, sunt "incomode" de atins, intervenția craniosinostozei devine complicată și poate să nu ofere rezultatele dorite.


